 Pasar al contenido principal
Pasar al contenido principal
EMBRIOLOGÍA DEL OÍDO INTERNO
El oído externo y medio comparten origen embrionario a partir del sistema branquial; sin embargo, el desarrollo del oído interno es a partir del otocisto ectodérmico, pero de forma independiente al resto del oído. Esto explica por qué las malformaciones del oído interno suelen ser aisladas, mientras que las de oído externo y medio suelen presentarse de forma combinada.
El desarrollo del oído interno se produce a partir de la placoda ótica, que es un espesamiento del ectodermo cefálico a ambos lados del romboencéfalo. La cóclea comienza su desarrollo en torno a la tercera semana de gestación y se completa en la octava semana. Por otro lado, el vestíbulo está completamente desarrollado en la semana 11 y los canales semicirculares entre las semanas 19 y 22, siendo el primer canal semicircular en desarrollarse el superior, seguido del posterior y el lateral. La osificación del laberinto se completa en la semana 23 y el desarrollo del oído interno estará completo en la semana 26.
TIPOS
Las malformaciones de oído interno pueden ser membranosas, óseas o mixtas. Las anomalías que pueden diagnosticarse mediante estudios radiológicos se deben a defectos que se producen entre la cuarta y octava semana, afectan al laberinto óseo y representan el 20% de los casos de hipoacusia congénita (malformaciones óseas); mientras que las lesiones posteriores que afectan al epitelio sensorial, no se pueden ver en las pruebas de imagen del hueso temporal y constituyen el 80% de los casos de sordera congénita (malformaciones membranosas).
CLASIFICIACIÓN
Existe una gran variedad de malformaciones y esto ha conllevado a tener diversas clasificaciones recogidas en la literatura. Actualmente la clasificación más aceptada de forma universal es la propuesta por Sennaroglu (Figura 1) y es la que se muestra a continuación. Ésta se fundamenta en la embriogénesis y cada tipo de malformación es consecuencia de la detención del desarrollo en un momento determinado.
a) Malformaciones cocleares.
- Aplasia laberíntica completa (deformidad de Michel): ausencia total de todas las estructuras cocleares y vestibulares. El peñasco puede ser hipoplásico y la cápsula ótica hipoplásica o aplásica. En la mayoría de los casos el conducto auditivo interno (CAI) consta únicamente del canal facial óseo. Se produce por la detención del desarrollo de la plácoda ótica a principios de la tercera semana de gestación.
- Otocisto rudimentario: representación milimétrica incompleta de la cápsula ótica (de forma redondeada u ovoide) sin presentar CAI. Los canales semicirculares pueden estar presentes pero muy rudimentarios. Se trata de una anomalía que está entre la deformidad de Michel y la cavidad común. Con respecto a la deformidad de Michel, la diferencia es que en ésta no hay desarrollo del oído interno; mientras que con la cavidad común es que en ésta última hay un espacio quístico ovoide o redondeado en lugar de la cóclea, separado del vestíbulo y con comunicación con el tronco del encéfalo a través de los nervios del CAI. El otocisto está causado por la detención del desarrollo después de que haya desarrollado la vesícula ótica, es decir, de forma más tardía a la tercera semana de gestación.
- Aplasia coclear: se caracteriza por la ausencia completa de la cóclea. Esto hace que el recorrido del segmento laberíntico del nervio facial sea aberrante, estando desplazado anteriormente y, por tanto, ocupando la ubicación normal de la cóclea. Existen dos subtipos en función del sistema vestibular:
- Aplasia coclear con laberinto normal: El vestíbulo y los canales semicirculares se desarrollan normalmente.
- Aplasia coclear con vestíbulo dilatado: El vestíbulo y los canales semicirculares están dilatados. Es importante diferenciar esta malformación de la cavidad común.
- Cavidad común: hay una cavidad quística única, ovoide o redonda, que representa la cóclea y el vestíbulo (no hay diferencia entre ellos), por lo que teóricamente esta cavidad tiene estructuras neurales cocleares y vestibulares. Puede haber canales semicirculares o partes rudimentarias de estos. El CAI habitualmente entra por el centro de la cavidad. Puede producirse por la detención del desarrollo en la cuarta semana de gestación.
- Hipoplasia coclear: la cóclea y el vestíbulo están separados entre sí (existe una buena diferenciación entre ambos), pero sus dimensiones son más pequeñas de lo normal. Existen diferentes subtipos:
- Tipo I (cóclea en forma de “yema”): la cóclea es como un pequeño brote que surge del CAI, esto es, como una cóclea pequeña en forma de yema sin arquitectura interna (no se pueden identificar modiolo ni septo interescalar).
- Tipo II (cóclea hipoplasia quística): la cóclea es más pequeña en cuanto a sus dimensiones pero su arquitectura externa es normal. El modiolo y septo interescalar están presentes pero defectuosos, aunque también puede existir una ausencia completa de modiolo creando una conexión amplia con el CAI. El acueducto vestibular y el vestíbulo pueden estar dilatados.
- Tipo III (cóclea con menos de dos vueltas): la cóclea tiene un modiolo más corto y la longitud del septo interescalar es más pequeña, lo que da lugar a un menor número de vueltas (menos de dos vueltas). La arquitectura tanto interna (modiolo y septo intercalar) como externa es similar a la de una cóclea normal, con menor número de vueltas y dimensiones más pequeñas. El vestíbulo y los canales semicirculares suelen ser hipoplásicos.
- Tipo IV (cóclea con espiras media y apical hipoplásicas): la cóclea tiene una espira basal normal, pero la media y apical son hipoplásicas y se ubican en una posición anterior y medial en lugar de su posición central normal. El segmento laberíntico del nervio facial generalmente se localiza por delante de la cóclea en lugar de su posición habitual.
- Partición incompleta de la cóclea: anomalía en la que hay una clara diferenciación entre cóclea y vestíbulo, manteniendo dimensiones externas normales pero con diversos defectos de arquitectura interna. Hay tres tipos diferentes según el defecto en el modiolo y septo interescalar.
- Partición incompleta tipo I (PI-I): la cóclea no presenta modiolo y con frecuencia la lámina cribiforme entre cóclea y CAI está defectuosa, lo que da como resultado una apariencia quística. Esto se acompaña de un gran vestíbulo dilatado quístico.
- Partición incompleta tipo II (PI-II) o deformidad de Mondini: la parte apical del modiolo y los correspondientes septos interescalares están defectuosos, esto hace que la parte apical de la cóclea tenga aspecto quístico, ya que las vueltas media y apical se fusionan para formar un ápice quístico. Cuando se acompaña de un vestíbulo dilatado y un acueducto vestibular ensanchado (por dilatación del conducto y del saco endolinfático) constituyen la llamada triada de la deformidad de Mondini. No es correcto definir esta anomalía como una cóclea de 1,5 vueltas, esto solo debe utilizarse para la hipoplasia coclear.
- Partición incompleta tipo III (PI-III): tiene septos interescalares pero el modiolo está completamente ausente. La cóclea está localizada en el extremo lateral del CAI, que está ensanchado, en lugar de su posición anterolateral habitual; y ambos están ampliamente comunicados por una deficiencia completa de la lámina cribosa.

Figura 1. Representación esquemática de la cóclea normal y las malformaciones cocleares.
A: cóclea normal, sección modiolar, Mo = modiolo, CA = apertura coclear, B = espira basal, M = espira media, A = espira apical, puntas de flecha = septos interescalares.
B: cóclea normal, sección inferior que pasa por el nicho de la ventana redonda (RWN). Punta de flecha = septo interescalar entre las espiras media y apical.
C: aplasia coclear con vestíbulo normal.
D: aplasia coclear con vestíbulo dilatado.
E: Cavidad común.
F: PI-I.
G: PI-II.
H: PI-III.
I: Hipoplasia coclear tipo I.
J: Hipoplasia coclear tipo II.
K: Hipoplasia coclear tipo III.
Fuente: Sennaroglu L. Cochlear implantation in inner ear malformations - a review article. Cochlear Implants Int. 2010 Mar;11(1):4-41.
b) Malformaciones del laberinto posterior:
- Deformidad de Michel.
- Cavidad común.
- Vestíbulo ausente, hipoplásico o dilatado.
- Malformaciones de los canales semicirculares: Ausentes, hipoplásicos o dilatados.
- Acueducto vestibular dilatado: el punto medio entre el laberinto posterior y el opérculo es mayor de 1,5 mm, en presencia de una cóclea y vestíbulo normales. La diferencia entre esta anomalía y la PI-II es que la cóclea y el vestíbulo son completamente normales en la TC y RM.
b) Malformaciones del CAI: Ausente, hipoplásico/estrecho o dilatado.
c) Anormalidades de la apertura coclear: la apertura coclear es hipoplásica si el ancho es menor de 1,4 mm. Es aplásico cuando no hay canal o está completamente reemplazado por hueso. Las anomalías de la apertura coclear se pueden acompañar de un CAI estrecho en la TC (se considera estrecho si el ancho del punto medio del CAI es menor de 2,5 mm).
Las malformaciones del oído interno pueden estar asociadas a hipoacusia neurosensorial hereditaria y no hereditaria. De hecho, la etiología de estas malformaciones puede ser idiopática, relacionada con una alteración genética congénita o en respuesta a la exposición a un agente teratógeno. Aunque pueden ocurrir de forma aislada, alrededor del 30% de estos casos están relacionados con un síndrome específico.
Los pacientes que presentan malformaciones de oído interno de causa genética suelen tener anomalías bilaterales y simétricas y clínicamente suele manifestarse con una hipoacusia neurosensorial profunda, tal y como se verá en el apartado de manifestaciones clínicas.
En el caso de la aplasia laberíntica completa lo más frecuente es que sea bilateral, pero también se han recogido en la literatura casos unilaterales, como en el síndrome de Wildervanck. La única causa genética sindrómica de esta malformación es el síndrome LAMM (trastorno autosómico recesivo caracterizado por esta malformación de oído interno de forma bilateral, asociado a microtia y microdoncia), causado por mutaciones en el gen FGF3; aunque también se ha descrito en un paciente con síndrome de mutación HOXA1 (gen involucrado: HOXA1).
En la literatura no hay datos recogidos sobre mutaciones genéticas que causen otocistos rudimentarios.
Por otro lado, está descrito que las cavidades comunes no tienen un origen genético. Sin embargo, hay estudios en los que se encontró asociación entre este malformación y mutaciones autosómicas recesivas del gen HOXA1 en una variante del síndrome Bosley-Salih-Alorainy, mutación del gen ROR1 en una familia con hipoacusia neurosensorial no sindrómica autosómica recesiva congénita y existe descrito un caso de cavidad común en un paciente con síndrome de deleción 22q11.
En los estudios que hay publicados no se ha informado de mutaciones genéticas específicas responsables de aplasia coclear, pero en modelos animales se ha relacionado con anomalías en los genes PAX2 (involucrados en el desarrollo del oído interno).
En cuanto a las particiones incompletas, en el caso de la tipo I, tampoco se han recogido genes específicos y la teoría actual es una alteración en el riego vascular. La PI-II se ha detectado en pacientes con síndrome de Pendred, en los que está presente como parte de la triada de Mondini y es debido a mutaciones bialélicas en SLC26A4. Se han descrito mutaciones en este gen en pacientes con acueductos vestibulares dilatados, con o sin PI-II; además de presentarse el acueducto vestibular dilatado de forma aislada en mutaciones de FOXI1 (un gen en estrecha relación con SLC26A4) y KCNJ10. La PI-III se conoce también como sordera ligada al cromosoma X. Fue la primera malformación en relacionarse con una herencia específica. Actualmente se sabe que es causado por una mutación del gen POU3F4, involucrado en el desarrollo del tubo neural (incluido lo que se convierte en la región anterior del hipotálamo). Esto explica por qué se han identificado distrofias hipotalámicas en pacientes con PI-III.
La hipoplasia coclear se ha observado en el síndrome branquio-oto-renal y puede detectarse en pacientes con trastorno del espectro óculo-aurículo-vertebral. También en el síndrome de rotura cromosómica de Varsovia por alteraciones bialélicas en DDX11 y en el síndrome de Pallister-Hall (en el que presentan de forma concomitante lesiones tipo hematoma en el hipocampo). La hipoplasia coclear tipo III se ha identificado en el síndrome de Waardenburg por mutaciones en SOX10 y la tipo IV es una anomalía frecuente en las distrofias musculares que afectan al α-distroglicano (distroglicanopatías), entre las que se encuentran el síndrome de Walker-Warburg, enfermedad músculo-ojo-cerebro y distrofia muscular congénita de Fukuyama.
La displasia de canales semicirculares, aislada o asociada a anomalías cocleares, se describe en varios síndromes como el síndrome CHARGE, la trisomía 13 y la trisomía 18. La agenesia aislada de los conductos semicirculares posteriores, sin afectación de los laterales, es extremadamente rara, pero se ha detectado en pacientes con síndrome de Waardenburg tipo II. Por otro lado, anomalías de forma bilateral en los canales semicirculares posteriores con afectación frecuente también de los superiores, pero conservando los laterales normales, se ha descrito en el síndrome de Alagille, producido por mutaciones en el gen JAG1. También se han detectado anomalías en los canales semicirculares en el síndrome de rotura cromosómica de Varsovia.
Se estima que la prevalencia de las malformaciones cocleo-vestibulares es de un 20-40% de los pacientes con hipoacusia neurosensorial congénita. Por ello se atribuye que éstas se producen en 1 de cada 10.000 o 20.000 casos.
La mayoría de pacientes que presentan malformaciones de oído interno presentan una hipoacusia neurosensorial bilateral de severa a profunda. Desglosando cada una de las malformaciones, los hallazgos audiológicos son las siguientes:
- Aplasia laberíntica completa (deformidad de Michel): No se obtiene respuesta en las pruebas audiológicas o pueden mostrar una hipoacusia neurosensorial profunda en las frecuencias bajas, lo que debe aceptarse como estimulación vibrotáctil.
- Otocisto rudimentario: al igual que en la anomalía anterior, no hay respuestas o una pérdida profunda en las frecuencias bajas, que también corresponde a la estimulación vibrotáctil.
- Aplasia coclear: tampoco tienen respuestas auditivas, la única estimulación puede ser vibrotáctil.
- Cavidad común: Hipoacusia neurosensorial profunda.
- Hipoplasia coclear: pueden presentar una hipoacusia neurosensorial, conductiva o mixta, desde leve a profunda.
- Particiones incompletas:
- IP-I: hipoacusia neurosensorial severa a profunda.
- IP-II: suele manifestarse como una hipoacusia progresiva (que puede variar hasta el grado profundo y puede ser simetría o asimétrica), pero también puede aparecer de forma súbita.
- IP-III: puede presentar una hipoacusia mixta (el componente conductivo puede ser debido a una cápsula ótica delgada) o neurosensorial profunda.
- Acueducto vestibular dilatado. Los hallazgos audiológicos son similares a los de una IP-II.
- Anomalías de la apertura coclear. Suele estar presente una hipoacusia neurosensorial severa a profunda. Como la cóclea es normal, pueden tener otoemisiones acústicas presentes, pero no pasaría la prueba de PEATC-A.
Dado que la intervención temprana tiene un impacto positivo en los resultados del tratamiento, el diagnóstico precoz de las malformaciones de oído interno es muy importante para el manejo de los pacientes con hipoacusia.
Las dos pruebas de imagen indicadas para el estudio de las patologías de oído interno son la tomografía computarizada (TC) y la resonancia magnética nuclear (RM) (Figura 2).
La TC es el estudio radiológico de elección para valorar las estructuras óseas del oído. Debe obtenerse en cortes axiales y coronales. Nos permite valorar anomalías del nervio facial, el tamaño del CAI y la morfología del oído interno.
Mientras que la RM permite una mejor valoración de los tejidos blandos, como el laberinto membranoso, los nervios del CAI (como el VIII par craneal) y los líquidos cocleares. Para valorar los nervios en el CAI deben realizarse cortes sagitales oblicuos y perpendiculares a los nervios y al CAI, identificándose el nervio facial en el cuadrante anterosuperior, el nervio coclear en el anteroinferior y los nervios vestibulares superior e inferior en los cuadrantes posteriores.
El nervio coclear puede estar ausente en las malformaciones del oído interno y esto es una contraindicación para la cirugía de implante coclear, que debe ser descartado mediante la RM. Pero también hay que tener en cuenta que es importante valorar la respuesta audiológica del paciente antes de denegar una candidatura a implante coclear debido a la ausencia de este nervio en la RM ya que puede que tengan algunas fibras auditivas presentes.
Por tanto, estas dos pruebas desempeñan un papel crucial en la caracterización de las malformaciones del oído interno y en la valoración de las estructuras anatómicas que permitirán seleccionar el tratamiento y el abordaje idóneos. Por ello, en conjunto, ofrecen al otorrinolaringólogo la información prequirúrgica necesaria que permitirá la decisión terapéutica, el consejo a los padres, el probable pronóstico del implante coclear, la selección del abordaje quirúrgico y del implante idóneos y alertará de las variantes anatómicas y de las posibles complicaciones quirúrgicas.
Por otro lado, la radiología nos brinda información valiosa sobre la posición de los electrodos en el postoperatorio.
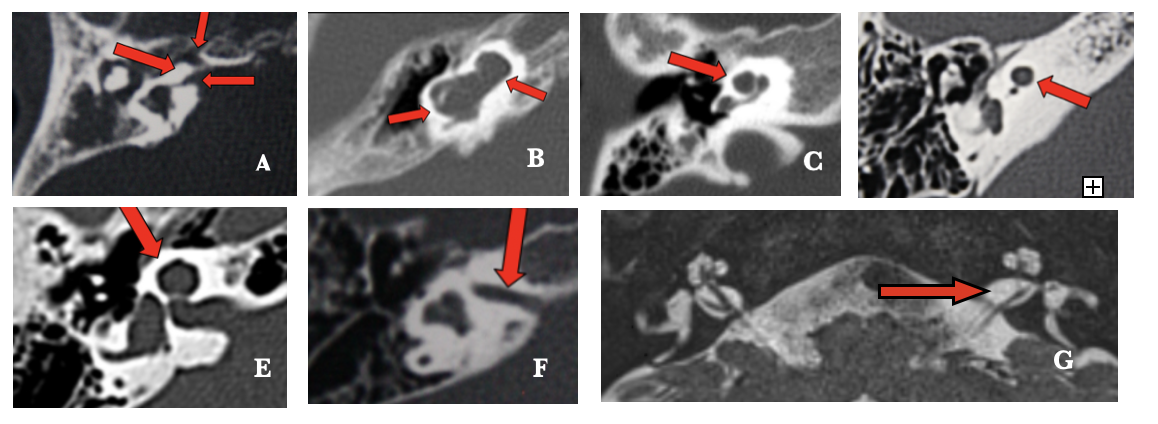 Figura 2. A: Aplasia coclear con vestíbulo dilatado. B: Partición incompleta tipo 1. C: Partición incompleta tipo II. D: Hipoplasia coclear tipo 1. E: Hipoplasia coclear tipo II. F: CAI estrecho. G: Aplasia del nervio coclear izquierdo con estenosis de la apertura coclear asociada.
Figura 2. A: Aplasia coclear con vestíbulo dilatado. B: Partición incompleta tipo 1. C: Partición incompleta tipo II. D: Hipoplasia coclear tipo 1. E: Hipoplasia coclear tipo II. F: CAI estrecho. G: Aplasia del nervio coclear izquierdo con estenosis de la apertura coclear asociada.
Tal y como se ha comentado anteriormente, es muy importante el diagnóstico precoz para conseguir una intervención temprana sobre la pérdida auditiva y evitar todas las consecuencias que tiene la misma en un niño durante el periodo de desarrollo.
Cuando comenzaron a realizarse los primeros implantes cocleares, las malformaciones de oído interno se consideraban como una contraindicación para la cirugía. Sin embargo, los avances en las técnicas quirúrgicas y en los dispositivos cocleares han permitido la implantación en cócleas malformadas; además de mejorar la inserción de la guía de electrodos, ya que al principio había mayor incidencia de inserciones incompletas debido a la longitud de los mismos. Con el desarrollo de electrodos más cortos se ha visto aumentada la frecuencia de conseguir una inserción completa de los mismos.
Fue en el año 1983 cuando se informó del primer caso de implante coclear realizado en una malformación de oído interno con resultado exitoso.
En los años noventa comenzaron a aparecer en la literatura pequeñas series de casos, hasta la actualidad en la que la cirugía de implante coclear en las malformaciones de oído interno se acepta como un abordaje quirúrgico estándar utilizando la vía del receso facial; teniendo en cuenta que los principales problemas que se pueden encontrar en la intervención son la anomalía del nervio facial, anatomía inusual del promontorio y ventana redonda, hipoplasia del oído medio, hiperneumatización u obliteración ósea completa de la mastoides. Todo esto puede dificultar o impedir por completo el acceso a la ventana redonda o al lugar de realización de la cocleostomía. Para evitar estas anomalías se han propuesto alternativas quirúrgicas con otros abordajes adicionales con el objetivo de exponer mejor el promontorio y el lugar de la cocleostomía.
En el caso de un nervio facial con un trayecto aberrante, como en la PI-I, cavidad común e hipoplasia coclear, el segmento vertical del nervio facial suele estar desplazado anteromedialmente hacia el promontorio y, por lo general, puede encontrarse sobre las ventanas oval y redonda. Esto hace que no pueda utilizarse como abordaje el del receso facial y como alternativas se disponen de la vía transcanal o la laberintotomía transmastoidea. Ésta última se utiliza sobre todo en la cavidad común e hipoplasia coclear, en las que el promontorio no está completamente desarrollado y no se produce el relieve característico del mismo. Con esto se consigue reducir el riesgo de lesión de nervio facial.
Por otro lado, la petrosectomía subtotal puede ser útil en algunas anomalías y se ha descrito en casos de hipoplasia coclear en forma de “yema” el abordaje a través de la ventana oval, retirando el estribo e insertando el electrodo a través de esta ventana en el vestíbulo.
Otro problema importante que puede surgir en la cirugía es la aparición de un gusher, que es una complicación que se produce al abrir la ventana redonda en algunos pacientes y eso supone un mayor riesgo de desarrollar meningitis postoperatoria. Se trata elevando la cabeza del paciente para disminuir la presión intracraneal y taponando la cocleostomía/ventana redonda. Para ello se han desarrollado electrodos especiales como se mostrará más adelante. Las cavidades comunes tienen una mayor asociación de anomalías de la pared lateral del CAI y, por tanto, mayor riesgo de gusher. Además puede precisarse en situaciones de gusher incontrolable la realización de una petrosectomía subtotal, obliterando la cavidad mastoides con grasa abdominal y cerrando la trompa de Eustaquio.
A continuación se muestra el tratamiento rehabilitador de la audición en función del tipo de malformación:
- Aplasia laberíntica completa (deformidad de Michel): Son candidatos a implante de tronco cerebral, ya que la cirugía de implante coclear no es posible debido a que no hay desarrollo del oído interno.
- Otocisto rudimentario: es una indicación de implante de tronco cerebral.
- Aplasia coclear: la única opción quirúrgica es el implante de tronco cerebral.
- Cavidad común: Pueden ser subsidiarios de implante coclear. En el caso de que el desarrollo de la audición y el lenguaje sea insuficiente, puede plantearse el implante de tronco cerebral (indicación posible).
- Para la cirugía de implante coclear en este caso, se puede realizar un abordaje mediante laberintotomía transmastoidea eligiendo un electrodo recto (no perimodiolar), para tener una posición en la periferia de la cavidad con mejor contacto con el tejido neural. Beltrame describió un electrodo especial para este tipo de malformación con un extremo inactivo. Se crean dos aberturas de laberintotomía en el área del canal semicircular lateral separadas por 3-4 mm. La parte inactiva del extremo del electrodo se coloca en la laberintotomía superior y el resto de guía se emplaza a lo largo de la pared interna de la cavidad a través de la laberintotomía inferior.
- Hipoplasia coclear: las opciones dependen del grado de hipoacusia neurosensorial y van desde audífonos (en el caso de pérdidas auditivas leves a severas) hasta el implante de tronco cerebral (en el caso de deficiencia del nervio coclear). En el caso de los pacientes con hipoacusia severa-profunda es posible rehabilitarlos mediante el implante coclear, de hecho la mayoría de los pacientes con hipoplasia coclear presentan pérdida auditiva de grado severo a profundo. Para ello se debe elegir un electrodo fino y más corto, ya que el número de vueltas es menor y con escalas más estrechas. Durante la intervención quirúrgica es esperable encontrar una trayectoria aberrante del nervio facial, puede ser difícil identificar el promontorio y la ventana redonda a través del receso facial; tal y como se ha expuesto anteriormente. En estas situaciones puede ser necesario asociar un abordaje transcanal o realizar una laberintotomía transmastoidea. Si con este dispositivo el desarrollo auditivo y del lenguaje es limitado, se puede considerar la implantación de un implante de tronco cerebral en el lado contralateral.
- Además, en algunos casos de hipoplasia coclear tipo III y IV pueden beneficiarse de la estapedotomía, ya que pueden tener pérdida auditiva pura conductiva o mixta donde el componente transmisivo se produce por la fijación del estribo; por ello se pueden beneficiar de la cirugía del estribo o de la adaptación de audífonos.
- Es importante tener en cuenta que una posible complicación durante la cirugía en la hipoplasia de tipo II es la aparición de gusher y mala colocación de la guía de electrodos del implante coclear en el CAI, debido a que el modiolo puede estar completamente ausente, creando una amplia conexión con el CAI. Esto puede conllevar la aparición de meningitis recurrente. El gusher que se puede producir durante la cirugía de la hipoplasia de tipo II puede resolverse utilizando un electrodo especial que presenta un “tapón” o “sello” cónico de silicona que permite un sellado eficaz de la cocleostomía.
- Particiones incompletas: los pacientes que presentan PI-I suelen ser candidatos a implante coclear (salvo que haya aplasia del nervio coclear, que en ese caso serían candidatos a implante de tronco cerebral), mientras que los pacientes con PI-II pueden necesitar al principio audífonos hasta que la progresión de la pérdida auditiva pueda hacerles candidatos a implante coclear. Los pacientes con PI-III que presentan hipoacusia neurosensorial o mixta de moderada a severa pueden tratarse con audífonos, mientras que cuando sea severa a profunda serán candidatos a implante coclear.
- A la hora de elegir el electrodo en la de tipo I y III se prefiere uno recto, no perimodiolar, mientras que en la tipo II se puede elegir cualquier electrodo. Siempre teniendo en cuenta el electrodo expuesto anteriormente con el “tapón” de silicona para sellar la cocleostomía. Como posible complicaciones en la cirugía pueden aparecer una fístula espontánea de líquido cefalorraquídeo y la meningitis recurrente, que puede verse tanto en PI-I (característico) como en PI-II (menos frecuente) debido, respectivamente, a un defecto en el área cribosa entre la cóclea y el CAI, a una base del estribo defectuosa y anomalías del modiolo. Por otro lado, los casos de PI-III siempre tienen un gusher de LCR de alto volumen durante la cirugía de implante coclear, pero la meningitis es muy rara ya que la platina del estribo es normal.
- Acueducto vestibular dilatado: el manejo quirúrgico es similar al de la PI-II.
- Anomalías de la apertura coclear: en estos pacientes los audífonos habitualmente no dan suficiente amplificación en pacientes cuando presentan una apertura coclear hipoplásica o aplásica. En los pacientes con apertura coclear hipoplásica bilateral con nervio coclear hipoplásico, es necesario probar una adaptación audiprotésica pero si no aportan suficiente audición funcional, estos pacientes normalmente llegan a ser candidatos a implante coclear; siendo necesario el implante de tronco cerebral si no proporciona suficiente audición funcional. En el caso de apertura coclear aplásica, está indicado el implante de tronco cerebral como terapia de primera línea.
Finalmente es fundamental realizar una correcta rehabilitación logopédica del lenguaje y comunicación.
Los resultados de la implantación coclear en malformaciones de oído interno que están recogidos en la literatura muestran un claro beneficio de la misma. Está descrito que experimentan una mejoría estadísticamente significativa del rendimiento del habla después de la implantación, independientemente del tipo o de la gravedad de la malformación. Se ha observado que los resultados son mejores cuando la lesión durante el desarrollo embriológico es más tardía. Por otro lado, por ejemplo, entre los pacientes con cavidad común y con hipoplasia coclear no se han encontrado diferencias estadísticamente significativas con respecto a la percepción del habla.
En cuanto a los parámetros de ajuste de programación del implante coclear se ha comprobado que los pacientes con malformaciones de oído interno necesitan valores de ancho de pulso y del nivel umbral de estimulación eléctrica y el nivel de confort más altos que los usuarios de implante coclear con cócleas anatómicamente normales. El defecto de las estructuras cocleares y el menor número de neuronas en el ganglio espiral de una cóclea malformada, hace que la percepción del volumen deseado pueda lograrse con niveles más altos en los parámetros de percepción de la sonoridad de la señal. Además se debe tener en cuenta que precisan revisiones más frecuentes parar realizar ajustes en los mapas de programación.
- Brotto D, Sorrentino F, Cenedese R et al. Genetics of Inner Ear Malformations: A Review. Audiol. Res. 2021, 11, 524–536. https://doi.org/10.3390/audiolres11040047.
- Costa R, Sennaroglu L, Uchiyama M et al. Histopathology of Inner Ear Malformations: Potential Pitfalls for Cochlear Implantation. Otol Neurotol. 2019 Sep;40(8):e839-e846. doi: 10.1097/MAO.0000000000002356.
- Farhood Z, Nguyen SA, Miller SC et al. Cochlear Implantation in Inner Ear Malformations: Systematic Review of Speech Perception Outcomes and Intraoperative Findings. Otolaryngol Head Neck Surg. 2017 May;156(5):783-793. doi: 10.1177/0194599817696502.
- Feraco P, Picccinini S, Gagliardo C. Imaging of inner ear malformations: a primer for radiologists. Radiol Med. 2021 Oct;126(10):1282-1295. doi: 10.1007/s11547-021-01387-z.
- Kocabay A, Cicek Cinar B, Ozbal Batuk M, Yarali M, Sennaroglu. Pediatric cochlear implant fitting parameters in inner ear malformation: Is it same with normal cochlea? Int J Pediatric Otorhinolaryngol. 2022 Apr;155:111084. doi: 10.1016/j.ijporl.2022.111084.
- Mazón M, Pont E, Montoya-Filardi A, et al. Malformaciones del oído interno: una aproximación diagnóstico práctica. Radiología. 2017;59(4):297-305. Doi: 10.1016/j.rx.2016.09.009.
- Ramírez LV, Cano HD, Lubinus FG. Malformaciones congénitas del oído interno. Revisión del tema. Rev. Color. Radiol. 2018; 29(3):4481-5.
- Sennaroglu L, Saatci I. A new classification for cochleovestibular malformations. Laryngoscope. 2002 Dec;112(12):2230-41. doi: 10.1097/00005537-200212000-00019.
- Sennaroglu L, Sarac S, Ergin T. Surgical Results of Cochlear Implantation in Malformed Cochlea. Otol Neurotol. 2006 Aug;27(5):615-23. doi: 10.1097/01.mao.0000224090.94882.b4.
- Sennaroglu L. Cochlear implantation in inner ear malformations - a review article. Cochlear Implants Int. 2010 Mar;11(1):4-41. doi: 10.1002/cii.416.
- Sennaroglu L, Bajin M. Classification and Current Management of Inner Ear Malformations. Balkan Med J. 2017 Sep 29;34(5):397-411. doi: 10.4274/balkanmedj.2017.0367.