 Pasar al contenido principal
Pasar al contenido principal
Un síndrome (sd) es definido como un conjunto de síntomas que se presentan juntos y son característicos de una enfermedad o de un cuadro patológico determinado, en ocasiones, por la concurrencia de más de una enfermedad. Es imprescindible conocer los síntomas que caracterizan los síndromes más comunes de la práctica diaria.
HENDIDURA PALATINA.
Las fisuras de labio, alveolo y paladar precisan de un estudio multidisciplinar por parte del ORL, el cirujano maxilofacial y el logopeda. Las malformaciones que afectan al paladar blando y al paladar óseo, afectan a la funcionalidad de la trompa de Eustaquio, interfiriendo con la aireación y el drenaje del oído.
La formación del paladar primario comienza hacia el final de la 4ª semana con la formación de los procesos faciales. Al final de la 7ª semana la fusión del proceso nasal medial con el proceso maxilar, y la fusión de ambos procesos nasales completa la formación de las estructuras palatinas primarias. El defecto en la fusión de estos procesos dá lugar al labio leporino. El paladar secundario surge de las crestas palatinas de los procesos maxilares, se fusionan entre sí y con el paladar primario.
La alteración durante este proceso ocasiona la patología propia de la hendidura palatina. El cierre definitivo del paladar se produce una semana más tarde que la formación del labio superior, por ello las hendiduras se consideran entidades distintas. Actualmente se acepta que la aparición de labio leporino y de fisura palatina corresponde a un mecanismo de heterogenicidad.
HENDIDURA PALATINA:
La formación del paladar primario comienza hacia el final de la 4ª semana con la formación de los procesos faciales. Hacia el final de la 7ª semana la fusión del proceso nasal medial con el proceso maxilar, y la fusión de ambos procesos nasales completa la formación de las estructuras palatinas primarias. El defecto en la fusión de estos procesos ocasiona el labio leporino.
El paladar secundario surge de las crestas palatinas de los procesos maxilares, se fusionan entre sí y con el paladar primario. La alteración durante este proceso produce los hallazgos propios de la hendidura palatina. El cierre definitivo del paladar se produce una semana más tarde que la formación del labio superior, por ello las hendiduras se consideran entidades distintas.
Actualmente se acepta que la aparición de labio leporino y de fisura palatina corresponde a un mecanismo de heterogenicidad.
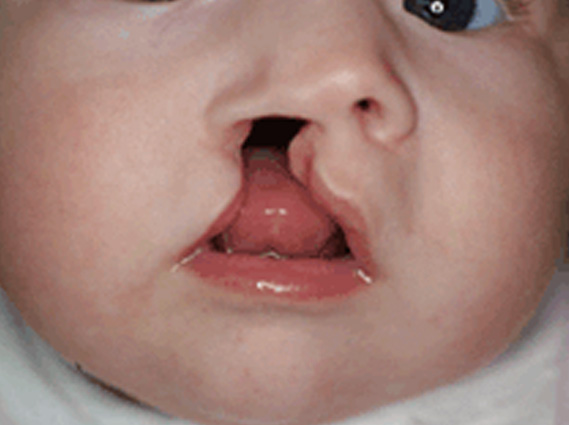
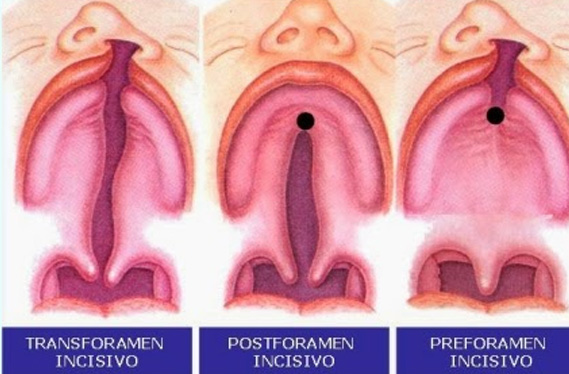
HENDIDURA PALATINA:
El diagnóstico de las fisuras de labio y paladar no sindrómicas se realiza por exclusión y supone una causa de herencia multifactorial. Aunque es difícil conocer la incidencia real se objetivó que los indios americanos tienen la mayor incidencia, 3.6:1000 nacimientos, seguidos por los japoneses, caucasianos y la raza negra (0.3:1000).
En cambio, el paladar hendido tiene la misma incidencia en todos los grupos raciales (1:2000 nacimientos). La relación varón: hembra es de 2:1 para fisuras asociadas de labio, paladar y alveolo, y de 1:2 para la fisura de paladar aislada. Las formas sindrómicas más conocidas y asociadas a paladar hendido (13-50%), son la secuencia de Pierre-Robin, síndrome De Treacher-Collins, trisomías 13 y 18, síndrome de Appert, síndromes de Stickler y Waardenburg.
El origen puede ser ambiental (alcoholismo, antiepilépiticos, radiación, virus…) o familiar (15% de los casos).
1. SINDROME DE DOWN
El sd de Down es el más común de los desórdenes cromosómicos y las secuelas en el área ORL son frecuentes. La mayoría de casos del síndrome se deben a una trisomía en el cromosoma 21, aunque una pequeña parte de se debe a una translocación, lo que da lugar a un alto grado de recurrencia.

La incidencia poblacional es de alrededor 1 de cada 650- 800 nacimientos vivos, y es de sobra conocida la relación entre la incidencia y la edad materna. El riesgo de Sd de Down en una mujer de 35 años es 1 de 385, en cambio en una mujer de 45 es de 1 de cada 28.
Características ORL: Orejas pequeñas. Canal auditivo externo estrecho. Aumento de la incidencia de OMS. Otoemisiones en ocasiones no productivas aunque la audición sea correcta. Cadena osicular alterada. Hipoacusia mixta (7%). Hipoacusia neurosensorial (8%). Obstrucción de vía aérea por canal nasal estrecho o hipertrofia adenoamigdalar. Macroglosia. Dificultad en la intubación por área subglótica estrecha. Riesgo de subluxación atlanto-occipital (15%), en procedimientos de intubación.
Características GENERALES: Fascies típica: braquicefalea, fisura palpebral, epicantus, nariz pequeña con dorso nasal deprimido. Retrognatia. Hipotonía. Laxitud articular. Líneas palmares marcadas. Alteraciones en el talón. Patología endocrina asociada (hipotiroidismo, leucemia). Patología digestiva asociada (atresia duodenal, enfermedad de Hirschprung) Patología cardíaca congénita.
2. ENFERMEDAD DE PIERRE - ROBIN.
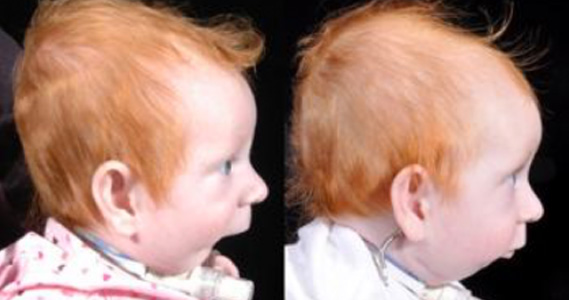
Se debe a un desorden embriológico caracterizado por la alteración del intrauterina del paladar y una hipoplasia mandíbular.
Características ORL: Paladar hendido. Retro o micrognatia. Intubación difícil. Apertura bucal pequeña. Proyección nasal marcada.
3. SINDROME DE TREACHER COLLINS.
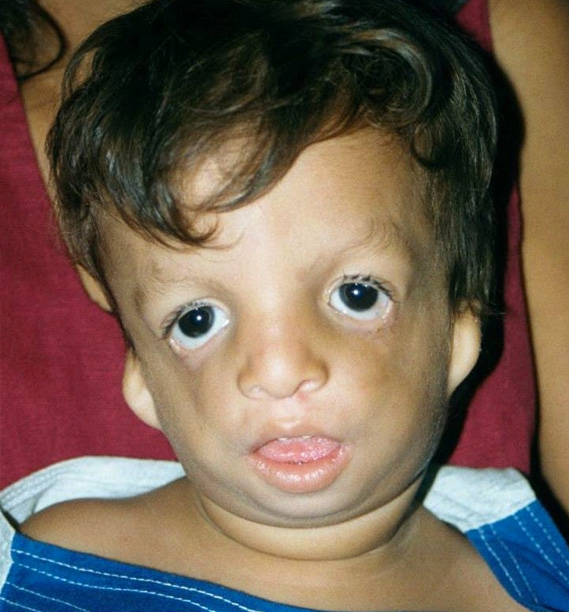
Es un síndrome caracterizado por una alteración de los arcos branquiales que lleva a una apariencia característica. La herencia es autosómica dominante y el único gen relacionado es el TCOF1. Una gran proporción de pacientes presentan nuevas mutaciones asociadas y una gran variabilidad.
Características ORL: Microtia y alteraciones del pabellón auricular. Fistula auricular. Hipoacusia frecuentemente conductiva debido a atresia de canal y asociada a malformación de cadena osicular. Paladar hendido. Micrognatia e intubación dificultosa. Hipoplasia mandibular. Alteraciones de la región malar. Nariz prominente. Nasofaringea estrecha. Atresia coana.
Características generales: fisura palpebral, coloboma.
4. SINDROME GOLDENHAR.

También conocido como síndrome oculo-auriculo-vertebral y se manifiesta como una microsomia hemifacial y alteraciones del primer y segundo arco branquial.
5. SINDROME DE CHARGE:
Caracterizado por Coloboma, patología cardíacada (Heart Disease), Atresia de coanas, Retraso en el crecimiento y desarrollo madurativo, alteración en Genitales y de oído (Ear defect) con hipoacusia en ocasiones asociada.
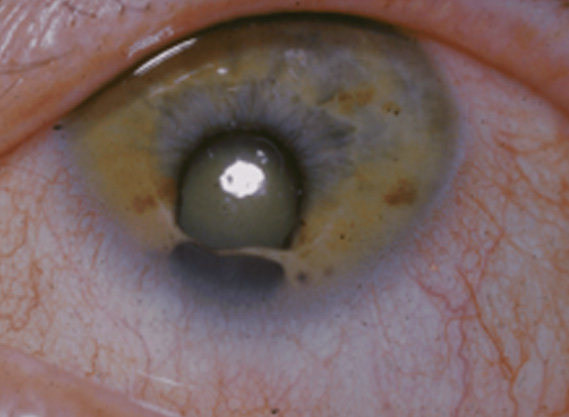
La causa es desconocida aunque coincide en la alteración del primer y segundo arco branquial, se han registrado algunos casos con herencia dominante, recesiva y varias anomalías cromosómicas (gen CHD7). Se han relacionado factores de riesgo como la diabetes materna, sangrados en los primeros meses del embarazo, talidomida, acido retinoico y las poblaciones que viven a gran altitud.
Características ORL: Microtia. Casi siempre unilateral y con una gran espectro de posibilidades, desde atresia de canal severa hasta grados menores. Alteraciones de la piel preauricular: quistes, fístulas… Hipoacusia mixta, conductiva o neurosensorial. Macrostomia.
Características generales: Asimetría facial. Quiste dermoide epibulbar. Defectos vertebrales. Alteraciones cardíacas. Alteraciones renales. Retraso mental.
6. SINDROME BRANQUIO-OTO-RENAL:
Caracterizado por una herencia autosomica dominante y por alteraciones a cualquier nivel del oído y displasia branquial y renal. Se conocen varias mutaciones en el gen EYA1 y en el gen SIX-1. La variabilidad clínica es amplia.
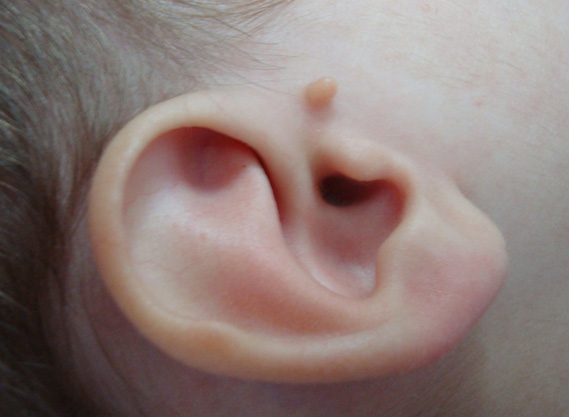
Características ORL: Alteraciones en oído: gran variabilidad de síntomas. Hipoacusia frecuentemente conductiva debido a alteraciones de la cadena osicular y también hipoacusia mixta. Las alteraciones del oído interno incluyen la displasia de Mondini y dilatación de acueducto vestibular. Fistula branquial o quiste asociado.
Característica general: malformación renal, hidronefrosis, duplicidad de sistema colector, displasia, agenesia renal uni o bilateral.
7. SINDROME POR DELECCION DEL 22Q11.2:
La delección del 22q11.2 también conocida como el sd velocardiofacial o sd de Di George, es una microdelección en dicho cromosoma y supone una herencia autosómica dominante. El fenotipo es muy variable pero en un 90% de los casos se objetiva una mutación de novo en el cromosoma 22.
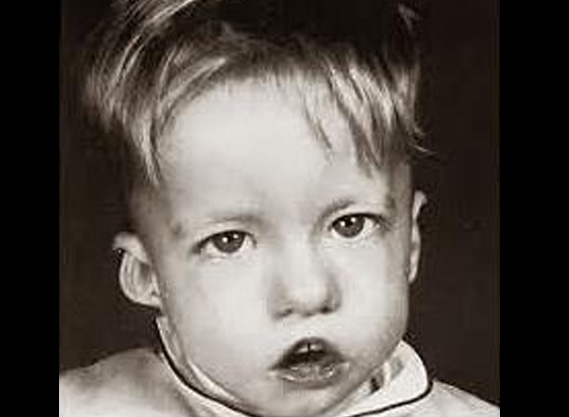
Características ORL: Paladar hendido, incompetencia del velo palatino y voz hipernasal. Pabellón auricular pequeño con hélix prominente y ausencia de lóbulo del oído, quiste preauricular, otitis media recurrente. Hipoacusia neurosensorial. Atresia de canal auditivo externo. Intubación dificultosa por obstrucción de vía aérea superior, membranas laríngeas y anillos vasculares, laringomalacia.
Características generales: Apariencia facial característica con una nariz prominente, cara alargada y nariz bulbosa. Patología congénita cardíaca. Retraso mental y de desarrollo. Estatura baja. Dificultades en la alimentación. Alteraciones psiquiátricas. Alteraciones inmunitarias. Disfunción de paratiroides con hipocalcemia.
8. CRANIOSINOSTOSIS:
Las alteraciones en el gen FGFR es común en todos los síndromes que producen craniosinostosis como es el caso del Sd de Crouzon, Apert, Muenke y Pfeiffer. Se transmiten mediante herencia autosómica dominante pero se han descrito nuevas mutaciones.
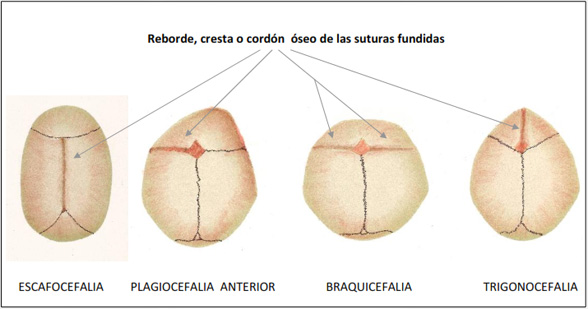
Características ORL: Dificultad en la intubación por obstrucción de la vía aérea. Alteraciones auriculares. Atresia de CAE. Alteraciones de oído medio con cadena osicular fija y disfunción de Trompa de Eustaquio. Hipoacusia neurosensorial.
Características generales: Craniosinostosis. Hipertelorismo. Proptosis. Sindactilia. Alteracion en pulgary dedos de los pies.
9. SINDROME DE ALPORT:
Caracterizado por la nefropatía (proteinuria y hematuria) e hipoacusia neurosensorial. Está causado por mutaciones en los genes del colágeno IV. La herencia observada es la ligada al cromosoma X afectando por ello más frecuentemente a los varones.
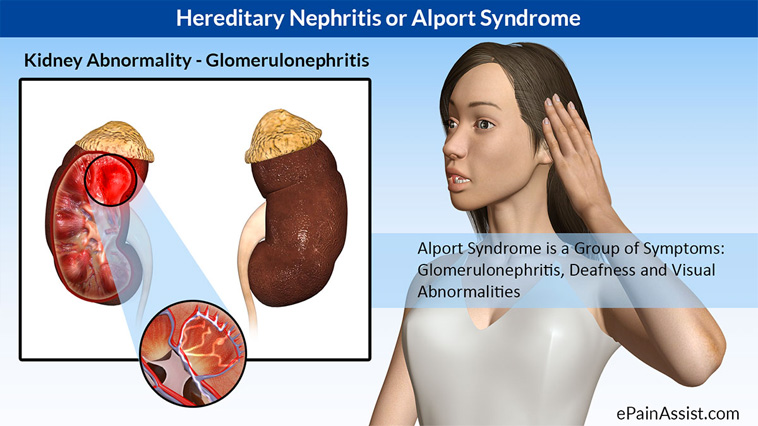
Características ORL: Hipoacusia neurosensorial.
Características generales: Defecto renal desde la infancia. Defecto ocular, lenticono y manchas maculares.
10. SINDROME DE PENDRED:
Caracterizado por la suma de hipoacusia congénita y alteraciones tiroideas debido a una mutación en el gen PDS (7 q31). Algunos autores defienden que es la forma sindrómica de hipoacusia congénita más frecuente. La mayoría de pacientes presentan hipoacusia bilateral moderada profunda y bocio eutiroideo.
Para tener un diagnóstico de verteza debe realizarse el test de perclorato. Es importante recordar que muchos de los pacientes presentan además la displasia de Mondini y el alargamiento del acueducto vestibular.
11. SINDROME DE USHER:
Caracterizado por hipoacusia neurosensorial y retinitis pigmentosa progresiva.
Es una de las causas autosómicas recesivas más frecuentemente ligadas a la hipoacusia y se clasifican en tres tipos dependiendo de la afectación otológica.
Tipo 1: Cofosis congénita. Ausencia de función vestibular. Los pacientes pueden ser candidatos a implante coclear. Retinitis Pigmetosa: Visión en túnel .
Tipo 2: Hipoacusia congénita con afectación predominante en las frecuencias agudas. Función vestibular correcta. Retinitis Pigmentosa: menos invalidante que el tipo 1.
Tipo 3: Hipoacusia progesiva. Funcion vestibular correcta o ausente. Retinitis Pigmentosa: tardía y variable dependiendo de la edad.
12. SINDROME DE ÄLSTROM:
Es un sd autosómico recesivo causado por la mutación ALMS1 y con una considerable variabilidad clínica.
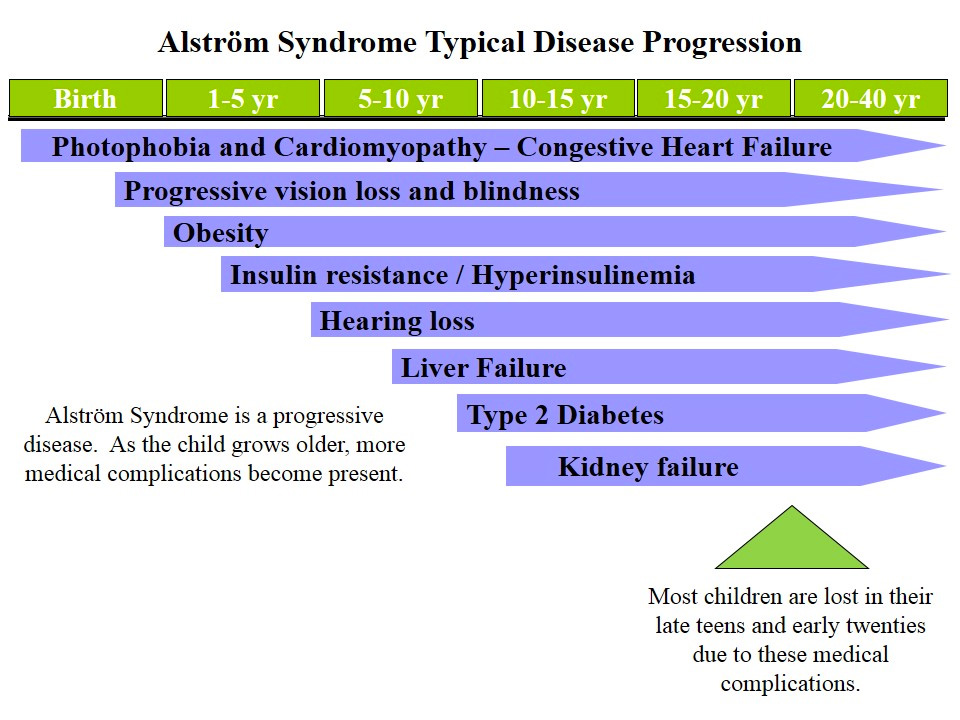
Características ORL: Progresiva hipoacusia a partir de la segunda década de la vida.
Características generales: Deterior progresivo de la retina. Fotofobia y nistagmus. Distrofia cónica. Obesidad. Diabetes tipo 2. Alteraciones renales. Diabetes y obesidad. Cardiomiopatía. Alteraciones endocrinas.
13. ACONDROPLASIA.
Supone la causa más frecuente de estatura corta desproporcionada que se caracteriza por una herencia autosómica dominante (gen FGFR3).
Se ha relacionado un aumento de la incidencia con la edad paterna y l 80% de los pacientes presentan mutaciones de novo.

Características ORL: SAHS o apnea central. Hipertrofia adenoamigdalar. Nasofaringe estrecha. OMA y OMS recurrentes. Hipoacusia mixta. Hipoplasia de tercio medio facial.
Características generales: Estatura baja. Rizomielia. Alteración de la tibia. Cifosis toracolumbar. Lordosis lumbar exagerada. Cráneo alargado y frente amplia. Tórax pequeño con compromiso respiratorio en algunos casos. Desarrollo neurológico: estenosis de canal cervical que puede llevar a muerte súbita en procedimientos como la intubación.
14. SINDROME DE BECKWITH- WIEDEMANN:
Es una alteración por sobrecrecmiento de algunas estructuras causado por alteraciones en el gen 11q15.
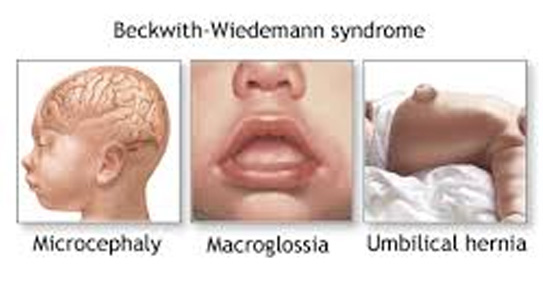
Características ORL: Macroglosia, supone un evidente riesgo anestésico. Alteraciones en el pabellón auricular.
Características generales: Macrosomia, visceromegalia, estatura alta. Hipoglicemia neonatal. Omfalocele. Riesgo aumentado de tumor embrionario: hepatoblastoma, neuroblastoma, rabdomiosarcoma. Alteraciones renales.
15. NEUROFIBROMATOSIS TIPO 2:
Es una entidad de herencia autosómica dominante y caracterizada por la presencia de schwanomas vestibulares. Se han objetivado nuevas mutaciones en el gen NF2 en un 50 % de los casos.
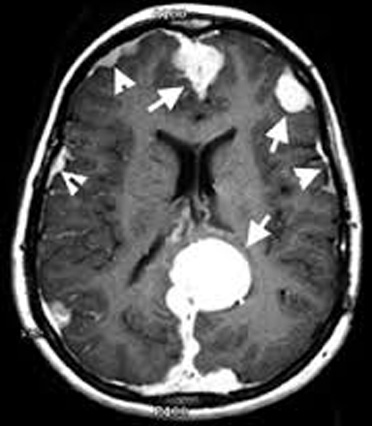
Criterios diagnósticos: Sólo un criterio se necesita para hacer el diagnóstico:
- Schwanomas bilaterales del VIII par craneal diagnosticados en la RM o el TC (biopsia no necesaria)
- Familiar de primer grado con NF2 y: a) schwanoma unilateral del VIIIpc, de inicio temprano (edad o 30 años) b) dos de los siguientes: Meningioma Glioma Schwanoma Menores con opacidad lenticular subcapsular posterior (catarata cortical juvenil).
- Schwanoma de VIIIpc unilateral diagnosticado por TAC o RNM de inicio precoz (detectado en paciente menor de 30 años) y 2 de los siguientes: a) Meningioma b) Glioma c) Schwanoma d) Catarata cortical juvenil
- Meningiomas múltiples y: a) schwanoma del VIIIpc unilateral b) 2 de los siguientes: Glioma, Schwanoma, Catarata cortical juvenil.
Características ORL: Hipoacusia, acúfenos, vértigo, parálisis facial causado de forma directa por el Schwanoma.
Características generales: Tumores de SNC: meningioma, ependimoma, tumor espinal, astrocitoma, schwanoma periférico. Manchas café-con-leche, menos comunes en la NF-1 Alteraciones oculares: cataratas, hamartromas retinianos.
16. SINDROME DE NOONAN:
Es un síndrome relativamente común de herencia autosómica dominante a causa de la mutación en el gen PTPN11 identificada en el 50% de los pacientes. EL gen KRAS también se ha relacionado en algunos casos.

Características ORL: Pabellón auricular ‘rotado’. Línea de implantación del cabello posterior baja. Hipoacusia variable. Micrognatia.
Características generales: Patología cardiaca congénita, estenosis pulmonar, cardiomiopatía hipertrófica. Retraso mental. Estatura baja. Alteraciones en el el esternón. Criptorquidia. Epicantus. Ptosis palpebral.
17. OSTEOGENESIS IMPERFECTA:
Se compone de un grupo de desórdenes hereditarios con los genes COL1A1 y COL1A2 como principales implicados en la alteración de la producción del colágeno tipo 1.
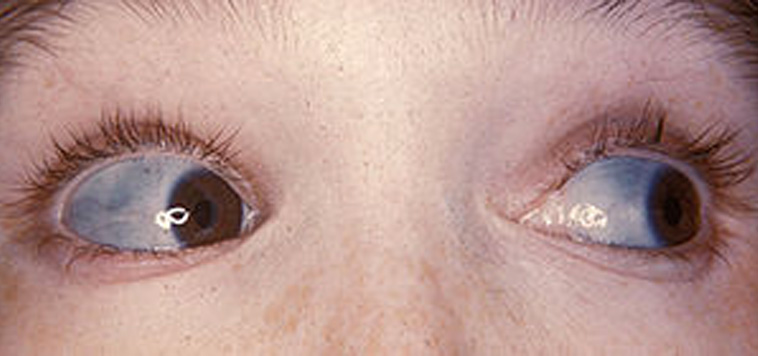
La característica principal son las fracturas patológicas y una hiperfragilidad del esqueleto. El tipo 1 es la forma más común y tiene una herencia autosómica dominante.
Características ORL: Hipoacusia neurosensorial.
Características generales: Esclera azul. Fracturas. Estatura baja.
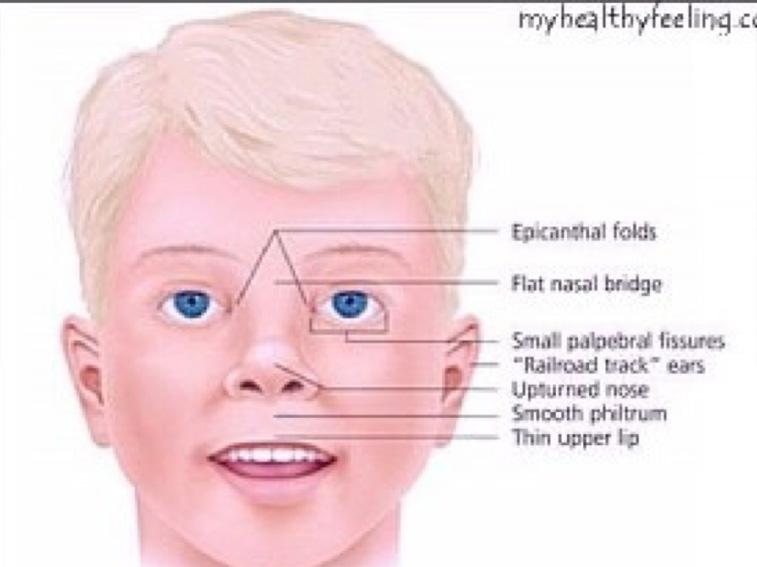
18. SINDROME DE PRADER WILLI:
Es un síndrome relativamente frecuente y destaca por la alteración en la alimentación y la obesidad de los pacientes.
Características ORL: SAHS.
Características generales: Hiperfagia y obesidad. Hipogonadismo. Retraso mental. Problemas de conducta. Estatura baja o alta para la edad. Hipotonia: en periodo neonatal.
19. SINDROME DE TURNER:
Sd causado con la ausencia parcial o total del cromosoma X en forma de mosaico en todas sus células. Tiene una frecuencia de 1 de cada 1800 niñas.
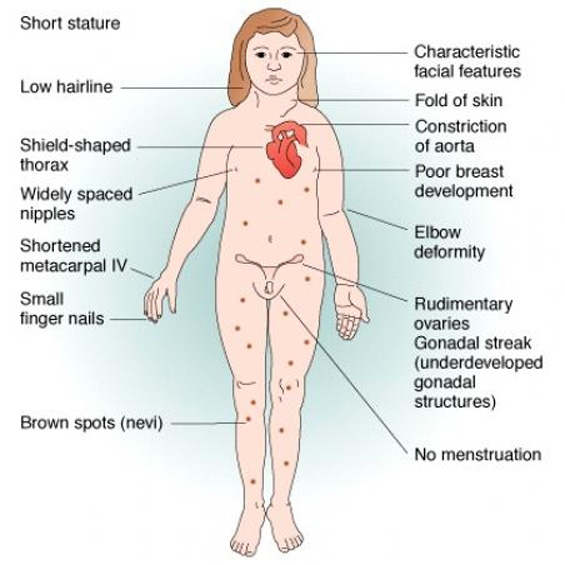
Características ORL: OMS recurrente. Hipoacusia neurosensorial. Cuello corto. Linea de implantación del pelo baja.
Características generales: Estatura baja. Disgenesia de ovarios. Coartación de aorta, válvula aórtica bicuspídea. Alteraciones renales. Dificultades de aprendizaje. Incidencia aumentada de alteraciones autoinmunes: tiroides, celiaquía, diabetes…
20. ENFERMEDAD DE WAARDENBURG:
Es el más común de las alteraciones sindrómicas con herencia autosómica dominante y se caracteriza por alteraciones pigmentarias y pérdida auditiva.
Los tipos 1 y 3 presentan alteraciones craniofaciales, y en el tipo 2 la apariencia es normal. En el tipo 4 se añade además la enfermedad de Hirschprung. Las mutaciones de los genes PAX3 ( tipos 1 y 3), MITF (tipo2), EDNRB, EDN3 y SOX10 ( tipo 4) son la causa del síndrome. Características ORL: Hipoacusia neurosensorial congénita. Ala nasal hipoplásica. Dorso nasal ancho. Cejas fusionadas.
Características generales: Heterocromia del iris. Canas prematuras. Hipopigmentación de la piel. Distopa canthorum. Alteraciones de los miembros. Enfermedad de Hirschprung.
21. SINDROME DE JERVELL Y LANGE-NIELSEN:
Es la forma homocigota de síndrome de QT-alargado con hipoacusia neurosensorial severa. La herencia es autosómica recesiva y causa mutaciones en los genes correspondientes a los canales de potasio (KCNQ1 y KCNE1).
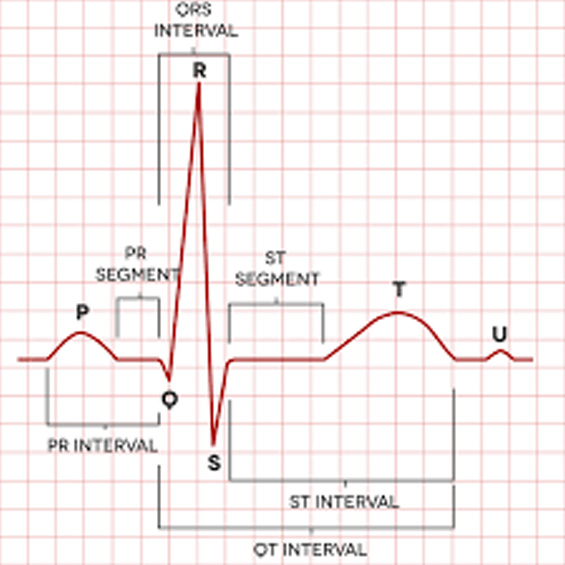
Características ORL: Hipoacusia neurosensorial congénita con ausencia de función vestibular.
Características generales: Intervalo QT alargado. Riesgo de Torsade de Pointes. Riesgo de fibrilación ventricular durante procedimientos anestésicos, ejercicio…
22. SINDROME DE GORLYN:
Es un síndrome autosomico recesivo que predispone a la aparición de carcinoma de células basales a consecuencia de una mutacion en el gen PTCH.
Características ORL: Quistes mandibulares. Paladar hendido y labio leporino.
Características generales: Carcinoma de células basales. Macrocefalia. Protuberancia frontal y biparietal. Hendiduras palmoplantares. Alteraciones oculares. Calcificación de la hoz cerebral. Alteraciones vertebrales.
23. HOLOPROSENCENFALIA:
Es un desorden heterogéneo y de tipo autosómico dominante. Se han relacionado genes como el SHH; SIX3, TGF1, ZIC2 y PTCH.
Características ORL: Alteraciones orales: desde labio leporino hasta frenillo lingual. Agenesia maxilar. Hipoplasia del tercio medio facial. Obstrucción de vía aérea. Alteraciones nasales.
Características generales: Alteraciones oculares: ciclopia. Retraso mental. Alteraciones de línea media: alteraciones cardíacas.
24. MUCOPOLISACARIDOSIS:
Poseen una herencia autosómica recesiva excepto el tipo II (sd de Hunter) con una herencia ligada al cromosoma X. Supone un grupo de alteraciones en el que cada entidad cursa con síntomas propios.

Características ORL: SAHS, intubación difícil. Hipoacusia neurosensorial. Subluxacion atlanto-axoidal. Macroglosia.
Características generales: Estatura baja. Alteraciones esqueléticas. Visceromegalia. Hepatoesplenomegalia. Debilidad articular progresiva. Alteraciones cardiacas. Retraso mental. Alteraciones corneales.
25. SINDROME ALCOHOLICO-FETAL:
Una exposición a la ingesta del alcohol durante el periodo intrauterino puede tener multiples efectos en el desarrollo del feto.
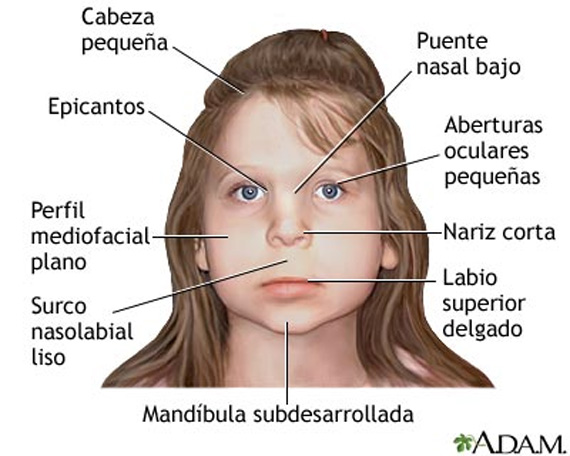
Caracteristicas ORL: Hipoacusia neurosensorial. Paladar hendido y labio leporino.
Caracerísticas generales: Peso bajo al nacer. Retraso de crecimiento. Retraso de desarrollo. Déficit de concentración. Fisura palpebral. Microcefalia.
26. CITOMEGALOVIRUS FETAL:
La severidad del virus dependerá del momento de la infección. Las secuelas durante el primer trimestres del embarazo son severas, en cambio la infección al final del embarazo con casi siempre asintomáticas.

Características: Hipoacusia neurosensorial. Microcefalia. Retraso de aprendizaje. Corioretinitis.
27. RUBEOLA FETAL:
La infección durante las primeras 1 - 6 semanas del embarazo tiene consecuencias severa para neonato.

Características: Hipoacusia neurosensorial. Retraso de crecimiento. Retraso mental. Alteraciones oftálmicas cataratas. Alteraciones cardiacas: ductus arterioso, estenosis de la arteria pulmonar.
HENDIDURA PALATINA.
Clasificación
Las clasificaciones existentes para tipificar las diferentes formas de fisuras labiopalatinas tienen como característica común describir los segmentos de la fisura que son afectados, sin considerar la severidad en que estos se encuentran distorsionados.
La severidad de la fisura es uno de los elementos más importantes a considerar en el planteamiento prequirúrgico de ésta.
Entre las clasificaciones tradicionales usadas para tipificar las fisuras labiopalatinas están la de Davis y Ritchie (1922), Veau (1931), Pfeiffer (1964), Kernahan (1971), Millard (1976) y Tessier (1979).
Todas estas hacen solo una descripción de los segmentos anatómicos involucrados en la fisura pero no cuan severamente está afectado. La clasificación de Kernahan tiene como alcance adicional un esquema donde se grafica de manera practica el tipo de fisura. Este es muy usado, sin embargo tampoco es específico en relación a la magnitud de la deficiencia de tejidos en la fisura.
Las clasificaciones como la de Mortier, considera la deficiencia de los tejidos sin embargo no considera el diámetro de la fisura labial y palatina.
Otra clasificación más reciente, es la de Ortiz -Posada, donde describe la severidad de la fisura (magnitud de la deficiencia de tejidos) considerando 3 componentes: nariz-labio, paladar primario y secundario.
Esta clasificación se basa en la deficiencia vertical y horizontal de los tejidos en la fisura considerando incluso aspectos bastante específicos como la integridad muscular, grosor del labio, profundidad del sulcus, etc. siendo detallada pero compleja y difícil .
La clasificación más utilizada es la propuesta por Kernahan y Starck, 1958. o Prepalatinas o paladar primario (por delante del agujero incisivo), con afectación de 1/3, 2/3 o completa. o Palatinas o paladar secundario, con afectación de 1/3, 2/3 o completa.

No hay que olvidar la clasificación propuesta por de VEAU y descrita en 1931: o Veau I: fisura de paladar blando aislada. o Veau II: fisura de paladar blando y duro. o Veau III: fisura unilateral completa de labio y paladar. o Veau IV: fisura bilateral completa de labio y paladar.
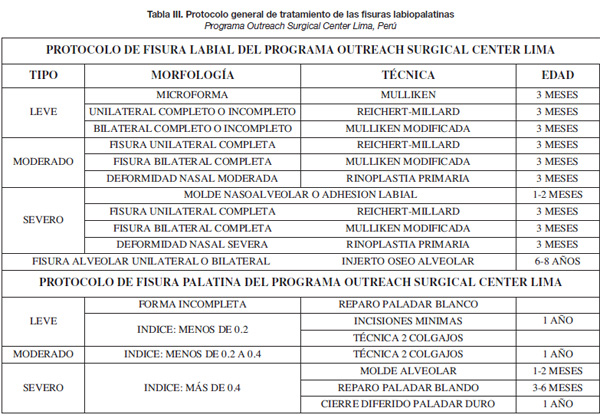
El diagnóstico se realiza por exclusión y tras una exploración completa.
HENDIDURA PALATINA.
A. Tratamiento quirúrgico:
Debe realizarse una reconstrucción funcional y estética.
La técnica de Veau- Wardill Kilner es la más utilizada, aunque en la técnica Furlow (doble Z-plastia) también parece que tiene buenos resultados. Se procura operar el paladar duro y blando antes de que se desarrolle el habla, antes de los 12 meses de edad o entre los 12 y 18 meses según el caso, siempre procurando la mínima lesión o secuela de la zona.
Posteriormente se debe perfeccionar el cierre mandicular con ortodoncia o cirugía ortognática.
La técnica para las alteraciones palatinas deberían seguir estos objetivos (Habbaby, 2000):
Cerrar la fisura palatina.
Prolongar el paladar o al menos evitar que se acorte en el postoperatorio.
Preservar la función normal del velo.
Lograr una competencia velofaríngea adecuada.
Producir un habla normal.
Permitir la función normal de las trompas de Eustaquio.
Conseguir un aparato masticatorio normal sin mala oclusión.
Interferencia mínima con el hueso subyacente.
No interferir con la fisiología nasal normal.
B. Tratamiento foniátrico:
El tratamiento foniátrico debe iniciarse lo más precozmente con el inicio del habla hacia los 2 años y hasta aproximadamente los 10 años, dependiendo de cada caso.
C. Tratamiento odontológico:
La prótesis “obturadora” permite una alimentación correcta, hábitos respiratorios normales, facilita la futura intervención quirúrgica y favorece el habla. El tratamiento ortodóncico es necesario si se está afectada la arcada dentaria o en casos de retraso en el crecimiento maxilar con maloclusión dentaria.
- Firth HV, Hurst JA, Hall J (eds) (2005) Oxford Desk Reference. Clinical Genetics. Oxford University Press, Oxford.
- Gorlin RJ, Cohen MM, Hennekam RCM (2001) Syndromes of Head and Neck, 4yh edn. Oxford University Press, Oxford.
- Winter R, Baraiser M (2004), London Dysmorphology Database Version 1.07.2004. Oxford University Press ( www.Imdatabase.com).
- Berkowitz S (1996) Cleft Lip and Palate- Perspectivwes in Management. Singular, San Diego.
- Shaw B, Semb G , Nelson P, et al (2001). The Eurocleft project. 1996-2001. IOS Press. Amsterdam.
- K.J Lee's . Essential Otolaryngology. Head and Neck Surgery. Ed Mc Graw- Hill 2016.